
Risk Management Plans Integration in Global Development Programs

Introduction
In global development programs, integrating Risk Management Plans has become a defining requirement for demonstrating patient safety, regulatory responsibility, and lifecycle oversight. The increasing complexity of therapies, expansion into multi-regional trials, and heightened regulatory expectations have pushed sponsors to adopt structured, evidence-driven risk strategies rather than fragmented regional approaches. As development teams transition from discovery to post-marketing phases, RMPs provide a unified framework for identifying, characterizing, and mitigating both known and potential risks.
This review synthesizes evolving regulatory expectations, global alignment challenges, and real-world examples to demonstrate how integrated RMPs shape the modern development landscape. By examining updates across EMA, FDA, MHRA, and PMDA, it explores how harmonized planning enhances safety, supports consistent benefit–risk reasoning, and strengthens regulatory confidence across regions.
Why RMPs Define Modern Development
RMPs have become central to development because they represent a shift from reactive safety management to proactive planning. Agencies worldwide now expect sponsors to document how risks are anticipated, what data supports risk characterization, and what mitigation measures ensure patient protection. EMA’s GVP Module V revisions emphasize early planning and measurable safety outcomes, while FDA’s REMS framework pushes for clear operational controls and monitoring effectiveness.
The global relevance of RMPs is evident in multi-regional submissions where inconsistencies in safety narratives can lead to review delays, additional data requests, or mandated mitigation measures. An integrated RMP provides a single source of safety truth, ensuring that messaging, rationale, and evidence remain aligned across regions. These expectations reinforce the role of RMPs as dynamic safety strategies rather than static documents.
Global Frameworks Converging
Although regulatory frameworks differ in terminology and structure, they increasingly converge around shared principles such as lifecycle assessment, documented reasoning, and measurable mitigation. EMA’s framework categorizes routine and additional risk minimization measures, whereas FDA focuses on enforceable elements, communication plans, and patient monitoring. MHRA, under the UK’s independent regulatory system, emphasizes transparent risk justification and post-authorization commitments, while PMDA integrates local clinical practice considerations into mitigation plans.
The convergence across 2024–2026 updates allows sponsors to develop a master RMP with adaptable regional sections. This unified design reduces duplication, improves consistency, and increases the clarity of benefit–risk narratives. For sponsors navigating accelerated development pathways, convergence also supports smoother regulatory communication and predictable approval timelines.
Global Comparison of Risk Management Frameworks
Regulatory Agency | Framework Name | Core Focus Areas | Risk Minimization Approach | Lifecycle Expectations |
EMA (Europe) | GVP Module V – Risk Management Systems | Structured identification of important risks & missing information; justification of safety concerns | Routine + additional risk minimization measures; communication tools; educational materials | Requires updates at approval, post-marketing, renewals; continuous benefit–risk evaluation |
FDA (USA) | Risk Evaluation and Mitigation Strategies (REMS) | Product-specific risk concerns that require enforceable controls | Medication Guides, Elements to Assure Safe Use (ETASU), implementation systems, monitoring of compliance | Periodic REMS assessment reports; evidence of mitigation effectiveness; modifications as needed |
MHRA (UK) | UK Pharmacovigilance Requirements (aligned with GVP) | Transparent safety justification, post-authorization commitments, signal evaluation | Uses EU-style RMM structure but with UK-specific expectations post-Brexit | RMP updates required at major variations, safety findings, renewals; strong post-marketing vigilance |
PMDA (Japan) | Japan RMP (J-RMP) | Integration of safety concerns with local clinical practice factors; post-authorization safety studies | Guides based on Japanese population data, additional monitoring, educational tools | Continuous updates reflecting local real-world data; close alignment with global RMP when available |
Embedding RMPs Early
Initiating RMP development early allows teams to define preliminary safety concerns, integrate enhanced monitoring, and align protocols with risk-based planning. When RMPs are embedded during Phase I or II, clinical and regulatory teams can anticipate uncertainties, ensure data collection methods address potential risks, and refine mitigation approaches as evidence evolves.
An example comes from an early-phase neurology trial where unexpected hepatic enzyme elevations were observed. Because the RMP framework was established early, the sponsor quickly implemented additional monitoring, modified inclusion criteria, and created a risk-based data review plan. Regulators acknowledged the proactive adjustments, enabling the program to progress without major delays.
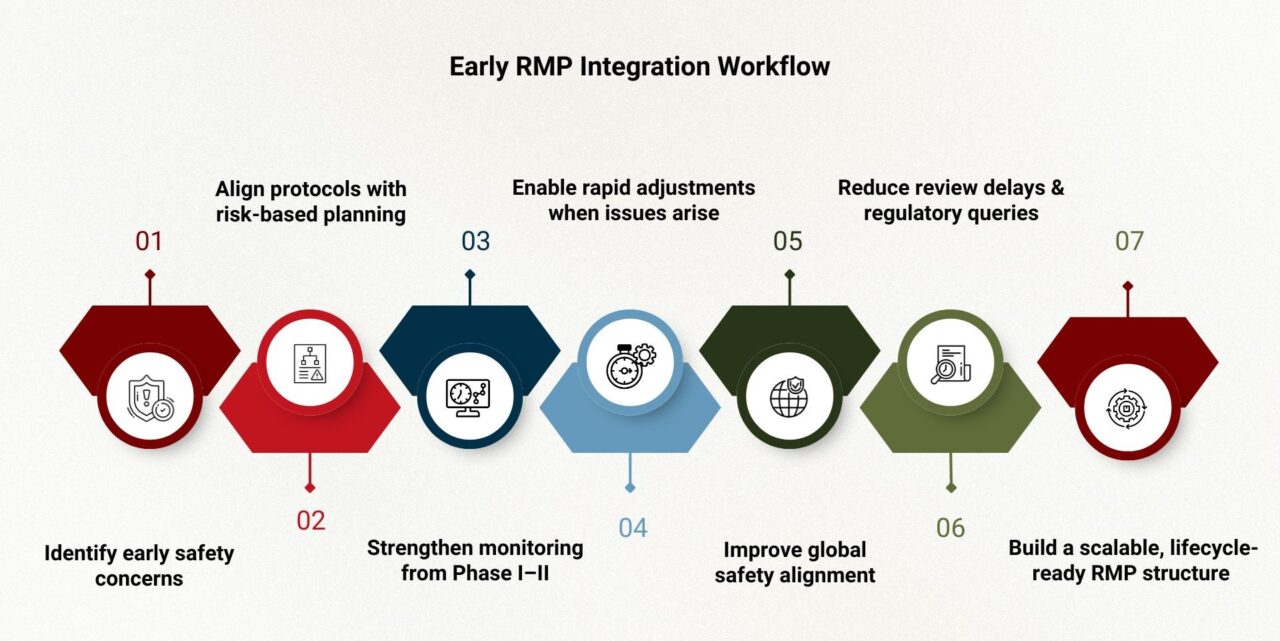
Turning Signals into Strategy
Safety signals gain significance only when translated into actionable risk strategies. RMPs provide the structure for interpreting signal data, evaluating causality, and determining whether additional mitigation is required. EMA’s recent updates emphasize the importance of demonstrating why a signal warrants action, while FDA expects clear documentation of how REMS elements address identified safety concerns.
Real-world examples illustrate this approach. A monoclonal antibody treatment displayed rare hypersensitivity reactions during early post-marketing use. The sponsor integrated additional educational materials, revised administration guidance, and instituted closer monitoring. Each decision was supported by evidence synthesis, which was documented in the updated RMP. This transparency strengthened regulatory confidence and demonstrated robust lifecycle oversight.
Evidence at a Glance
An integrated RMP stands on the strength and clarity of the evidence behind it. Regulators expect every identified or potential risk to reflect a transparent link between observed signals and the underlying data, whether it comes from early-phase clinical results, mechanistic insights, or emerging real-world patterns. EMA’s GVP Module V and FDA’s REMS guidance emphasize that unsupported assumptions weaken credibility, while structured evidence elevates the quality of the safety narrative. When sponsors present a clear, unified interpretation of data, regulators can more easily evaluate benefit–risk balance without seeking additional justification. This synthesis becomes even more essential in global submissions, where consistent evidence interpretation prevents conflicting messages between regions and supports smoother, faster reviews.
Lifecycle Oversight Driving Global Readiness
Lifecycle oversight transforms an RMP from a regulatory requirement into a dynamic safety tool. Regulators expect sponsors to continuously reassess risks as exposure expands and new safety insights appear not only at submission but throughout the product’s lifecycle. By updating the RMP as data evolves, sponsors demonstrate ongoing vigilance and adaptability, which agencies increasingly view as signs of operational maturity. Consistent oversight across regions also ensures that safety strategies remain aligned in multi-regional submissions, reducing requests for clarification and minimizing the risk of divergent regional commitments. Products supported by strong lifecycle oversight tend to move through renewals, extensions, and inspections with fewer hurdles, because regulators can clearly see how safety reasoning has evolved and how decisions were documented over time.
Documentation That Builds Trust
High-quality documentation is one of the most immediate indicators of scientific and regulatory reliability. In an RMP, documentation must convey not just what risks exist but why they are prioritized and how mitigation measures address them. Clear narratives that connect evidence, interpretation, and planned actions make it easier for reviewers to understand the sponsor’s safety logic. Equally important is alignment regulators expect the RMP to match information in CSRs, labeling proposals, IBs, and periodic safety reports. Even minor inconsistencies can undermine confidence or delay approval. When sponsors maintain consistent, transparent documentation across all phases including post-marketing updates regulators gain assurance that risks are monitored responsibly and communicated accurately, reinforcing trust throughout the product’s global lifecycle.
Conclusion
The integration of RMPs into global development programs reflects a broader shift toward evidence-driven, lifecycle-oriented safety planning. As frameworks converge across regulatory regions, sponsors must adopt unified, adaptive RMP architectures that support transparent decision-making and harmonized safety communication. The future of global development lies in predictive risk modeling, real-world data integration, and continuous evaluation of mitigation effectiveness. Products developed under such structured safety strategies will be better positioned to achieve regulatory approval and maintain strong post-marketing benefit–risk profiles.
References
Risk Management Plans — European Medicines Agency (EMA) – Official page on RMPs in the EU and links to guidance documents
https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/pharmacovigilance-marketing-authorisation/risk-management/risk-management-plans
EMA Good Pharmacovigilance Practices (GVP) — Central resource for the EU GVP framework (including links to Module V and other modules)
https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/pharmacovigilance-post-authorisation/good-pharmacovigilance-practices-gvp
Guidance on pharmacovigilance procedures — MHRA (U.K. Government)
https://www.gov.uk/government/publications/guidance-on-pharmacovigilance-procedures/guidance-on-pharmacovigilance-proceduresFDA Risk Evaluation and Mitigation Strategies (REMS) — U.S. FDA’s official safety framework for risk mitigation
https://www.fda.gov/drugs/drug-safety-and-availability/risk-evaluation-and-mitigation-strategies-rems
PMDA Safety Information and Pharmacovigilance (Japan) — Overview of post-marketing safety measures by Japan’s regulatory authority
https://www.pmda.go.jp/english/safety/info-services/0001.html
FAQs
What is a Risk Management Plan in global development programs?
Why is RMP integration important in multi-regional clinical trials?
How does real-world evidence help in Risk Management Plans?
What is the difference between RMP and REMS in global programs?
How often should sponsors update a Risk Management Plan?
Recent Posts
- A Sponsor’s Guide to Signal Detection for IND, NDA, BLA, and Post Marketing Programs June 22, 2026
- Risk Management Plans Integration in Global Development Programs March 31, 2026
- Post-Marketing Surveillance (PMS) in Regulatory Affairs: A Complete Guide March 24, 2026
- PBRER vs PSUR: Key Differences Explained March 16, 2026
- Global Signal Detection Trends Transforming Drug Safety 2026 March 4, 2026